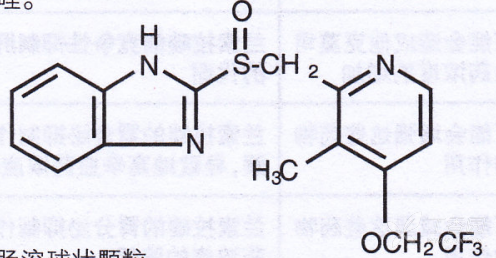
Lansoprazole Enteric-coated Capsules
请仔细阅读说明书并在医生的指导下使用
核准日期:2014年4月2日|修改日期:2015年11月16日
胃溃疡、十二指肠溃疡...登录
饭前服用。对于胃溃疡...登录
1. 警告胃癌对兰索...登录
网页版仅展示部分,【药物相互作用、不良反应、儿童用药等】使用微信扫一扫,在用药助手 App 看