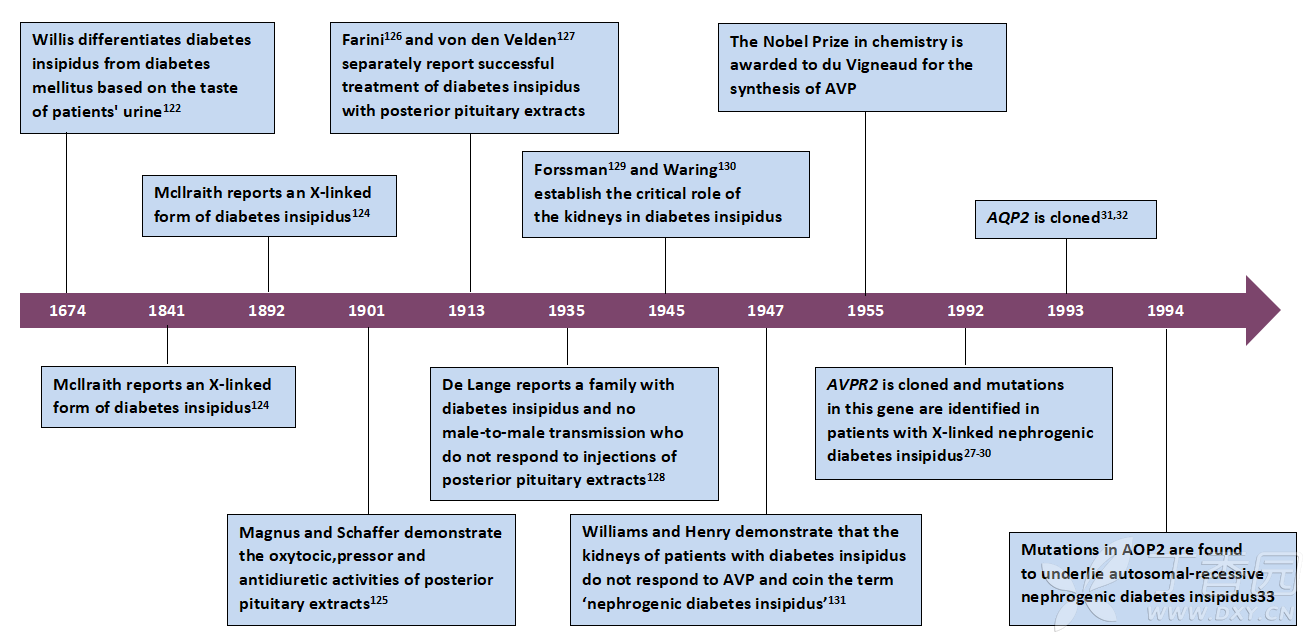
大约90%的遗传性先天性肾性尿崩症患者有AVPR2突变10-11。由于该基因位于X染色体上,因此AVPR2突变具有X连锁遗传模式,故而先天性肾性尿崩症患者多为男性。偶尔也会有因AVPR2突变导致先天性肾性尿崩症的女性患者8-9,12-15。在一项研究中,64例AVPR2突变的女性携带者中有16例(25%)表现出多尿症状,其中4例患者(6%)被诊断为完全性先天性肾性尿崩症,而不是下文所描述的较轻的部分性肾性尿崩症表型8。在一些发生AVPR2突变的女性携带者中,她们的症状由偏态X‑失活所引起,但是这些症状不一定与白细胞中观察到的X‑失活模式相关16。据推测,组织之间的X‑失活模式可能不同,因此白细胞中观察到的模式可能不能反映出肾脏中的模式。根据英国伦敦大奥蒙德街医院(Great Ormond Street Hospital)的经验,20个具有AVPR2突变的家系中,有2名女性亲属被诊断为完全性肾性尿崩症(D. Bockenhauer & D. Bichet,未发表的成果)。其中一名妇女是她家族中的先证者,但并不是所有的女性突变基因携带者都得到了系统性研究。在一个家系的另外2名女性突变基因携带者中,观察到了部分性肾性尿崩症的症状17。由于建立者效应,一些AVPR2突变被反复发现。迄今为止,已描述了超过300个家系中的超过250种不同的AVPR2突变情况1818,有趣的是,这些突变大多是错义突变,且当进行体外评估时,这些突变似乎可编码有功能性的、但却错误折叠的受体,这些受体在内质网(endoplasmic reticulum,ER)中被保留并降解19。
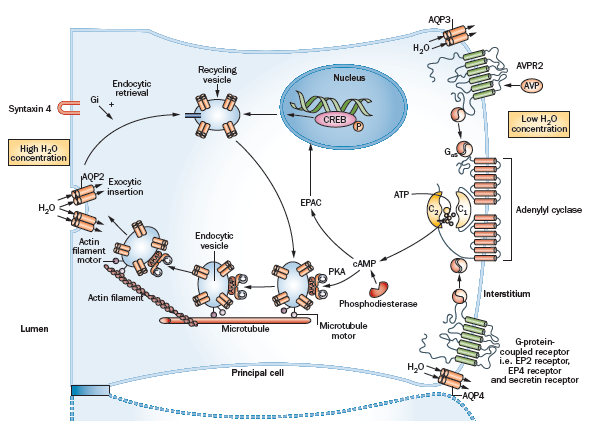
cAMP的作用既可通过经典的PKA/cAMP依赖性蛋白激酶途径介导,也可通过鸟嘌呤核苷酸交换因子如cAMP直接激活的交换蛋白(exchange protein directly activated by cAMP,EPAC)来介导59。与PKA一样,EPAC含有一个进化上保守的cAMP结合结构域,可感知细胞内cAMP水平,并充当分子开关控制多种生物学功能60。EPAC可能参与AQP2丰度的长期调控,而蛋白激酶A的调控具有独立的短期效应61。