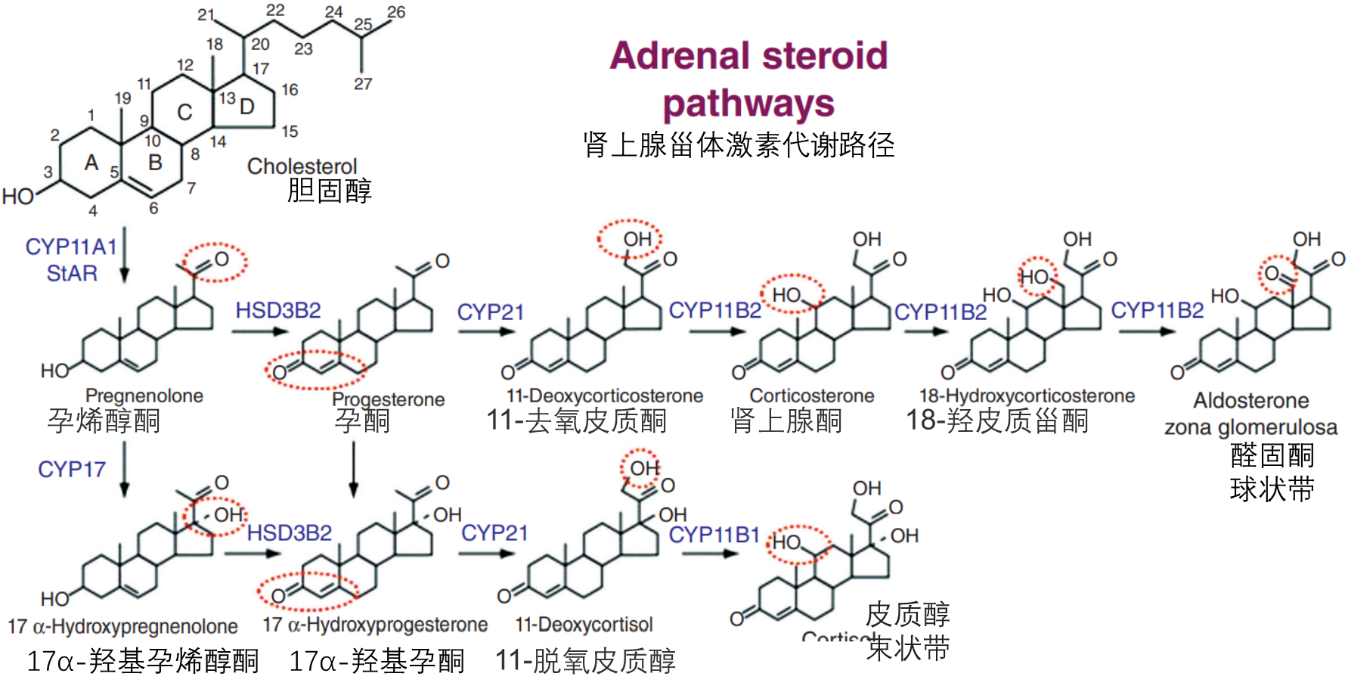
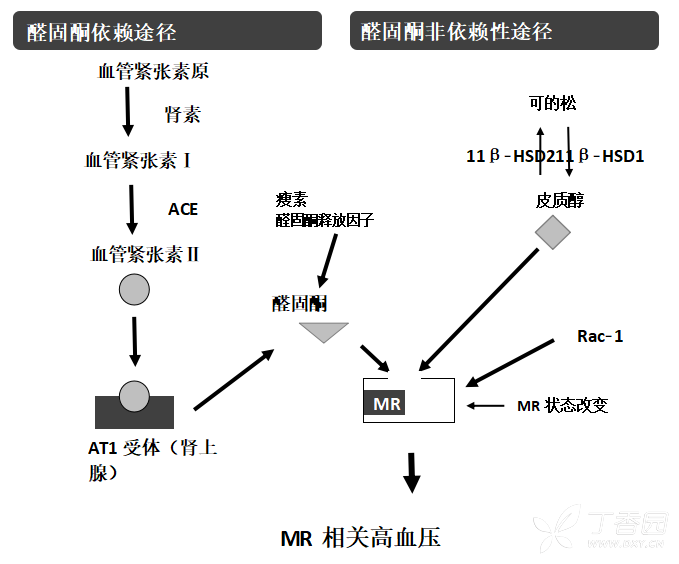
临床和生化证据表明原发性高血压和 PA 患者之间存在灰色区间,因此形成了连续疾病谱链假说,该疾病链包括原发性高血压、低肾素性高血压和双侧特发性醛固酮增多症(idiopathic hyperaldosteronism,IHA)[9-10]。与原发性高血压患者相比,醛固酮-肾素比值(aldosterone to renin ratio,ARR)和血浆醛固酮升高的患者更易出现顽固性高血压,即使不符合 PA 的诊断标准[11]。这种高血压被定义为“醛固酮相关高血压”,很可能是一种顽固性高血压。
2、肾素是由肾脏入球小动脉从肾素的前体,肾素原合成而来,是 RAAS 的关键调节因子[101]。肾素储存在储存颗粒中,并从储存颗粒中释放,催化血管紧张素原转化为血管紧张素 I。血管紧张素 I 通过肺中的血管紧张素转换酶转化为血管紧张素 II(angiotensin II,Ang II),肺中 ACE 的浓度最高。血管内皮细胞的管腔膜、肾脏的肾小球、肾上腺和大脑也可以形成 Ang II[102-103]。对于无肾脏的受试者,在其血浆中也能检测到低水平的 Ang II,这就可能是来自这些肾外来源[104]。血管内皮局部产生的 Ang II 可能有助于调节血管张力[105]。血管内皮局部产生的 Ang II 在维持血管张力方面也发挥着作用,这也有可能是促使高血压发生发展的一个因素[106]。
3、Ang II 的直接和间接作用导致小动脉血管收缩和钠水潴留。Ang II 的血管收缩作用可以是直接作用于血管平滑肌细胞或通过刺激交感神经系统间接作用。这些作用可纠正低血容量和低血压。Ang II 直接刺激早期近曲小管对钠的重吸收,并增加醛固酮的分泌[107-108]。来自肾上腺的全身和局部产生的 Ang II 都有助于醛固酮的产生[102]。Ang II 的作用是通过血管紧张素 1 型(angiotensin type 1,AT1)和 2 型(angiotensin type 2,AT2)受体介导的[109]。血管和肾小管的作用主要由 AT1 受体介导。AT2 受体功能没有很好的表征,但通常是与 AT1 受体在血管系统和肾脏中的作用相反。
4、Ang II 与肾上腺皮质球状带中的 I 型受体结合并导致磷脂酶 C 的上调。这是一种 G 蛋白偶联受体,可激活蛋白激酶 C,进而产生肌醇三磷酸和 1, 2 二酰基甘油[110]。这会增加细胞内钙的释放以及蛋白激酶和转录因子的激活[111]。这最终导致醛固酮合酶的激活和醛固酮的合成[112]。
5、Ang II 还可以通过 AT1 突触前受体激活交感神经系统。这导致精氨酸加压素释放增加、促肾上腺皮质激素(adrenocorticotropin,ACTH) 合成增加以及促甲状腺激素(thyroid stimulating hormone,TSH)和生长激素增加。在肾素分泌增加的情况下,例如肾动脉狭窄和与有效减少血浆容量(例如心力衰竭和肝硬化)相关的正常血压状态,血管紧张素 II 是 BP 的重要调节剂[113-114]。Ang II 的慢性激活导致血管重塑和血管系统僵硬度增加。
6、醛固酮作用由 RAAS 调节。醛固酮与 Ang II 和 1 型 Ang II 受体调节的途径相互作用[115]。醛固酮增加活性氧的形成,进而激活参与细胞生长、胶原形成和炎症的信号转导途径[116]。因此,Ang II 和醛固酮增强了彼此的作用。醛固酮还上调血管平滑肌细胞中的 1 型 Ang II 受体,从而导致血管收缩增强[117]。醛固酮和 Ang II 的协同作用会引发血管炎症和纤维化,并导致高血压。Framingham 等多个研究发现[118-119],血浆醛固酮和肾素分别与5年内收缩压的增加程度和高血压的发展呈正相关和负相关。生理范围内偏高的醛固酮水平可能导致高血压的发生;然而这需要进一步的研究来证实。
1、醛固酮生成增加是由钠缺乏和血浆容量减少触发的。这同样会触发肾脏释放肾素从而刺激 Ang II 的生成。钾在调节醛固酮方面也起着关键作用。钾在生理范围内的微量增加都会对醛固酮的合成产生显著影响。这表明肾上腺皮质球状带(zona glomerulosa,ZG)细胞对血浆中钾水平的微量变化具有敏感性[138]。钾和 Ang II 具有协同作用,因为 Ang II 诱导的醛固酮生成同时也受钾浓度的调节[139]。钾的增加会导致 ZG 细胞的细胞膜发生去极化。这进而导致电压依赖性 L 型和 T 型钙通道的开放,从而使细胞内的钙浓度迅速升高。钙的升高会激活钙调蛋白和 Ca2+-钙调蛋白依赖性蛋白激酶,使转录因子磷酸化,刺激细胞色素 p450(cytochrome p450,CYP)11β2 基因转录[140]。已得到证实,内脏脂肪细胞也可以分泌盐皮质激素释放因子,从而上调醛固酮合酶的表达并刺激醛固酮分泌[141]。