下载
App
App

专属顾问

返回
顶部
顶部
| 病因分类 | 具体病因 |
| 结构损害 | 血管因素:缺血性和出血性中风,血管畸形(如烟雾病) 肿瘤 脱髓鞘病变 |
| 内分泌和代谢性疾病 | 非酮症性高血糖 低血糖症 低钙血症 低钠血症或高钠血症 尿毒症 获得性肝脑变性 甲状腺功能亢进 甲状旁腺功能减退 /甲状旁腺功能亢进 |
| 感染性疾病 | 弓形虫病 HIV脑病 朊病毒病 |
| 药物因素导致 | 左旋多巴 可卡因 安非他命 抗惊厥药物 锂 抗胆碱能类 安定的撤药反应 |
| 自身免疫性/附肿瘤因素 | 小舞蹈病 风湿性疾病:系统性红斑狼疮、抗磷脂抗体综合征、系统性硬化症 自身免疫性神经综合征:抗CRMP-5, 抗NMDA, 抗Hu (ANNA-I), 抗Yo, 抗LGl1, 抗CASPR2, 抗GAD65, 抗lgLON5 |
| 其他 | 真性红细胞增多症 Postpump 舞蹈病 |
83岁女性,因异常运动18个月就诊。运动最初从她的手和脚开始,然后扩展到近端的胳膊和腿。睡眠期间无运动。此外,在过去的8个月里,她的家人注意到她变得易怒和开不恰当的玩笑。否认有记忆问题、抑郁或自杀意念。无明显的家族史。 检查显示中度全身性舞蹈病伴舞蹈步态。存在面部上方舞蹈症,也出现额肌和眉毛摆动。她表现出冲动性额叶去抑制症状,包括在考试期间开不恰当的玩话。存在扫视的启动延迟,在水平方向比垂直方向更突出,她需要通过眨眼或用力摇头来进行扫视。反扫视检查也存在异常。她也有运动无法持续的症状,包括无法维持伸出舌头和挤奶女工征阳性。存在悬吊抽搐,在股四头肌反射测试中表现为长时间伸膝。 她最初被诊断出患有“老年性舞蹈病”。全血细胞计数、甲状腺功能检查、血清钙和甲状旁腺激素均正常。脑部CT显示轻度至中度双侧尾状核萎缩。经过适当的遗传咨询后,进行了基因检测,发现HTT基因中有40个CAG片段重复。开始服用利培酮0.5mg/d以控制舞蹈病的症状。 |
点评 慢性病程(超过1年)提示舞蹈病为遗传性病因,其中成人中最常见的是亨廷顿舞蹈病(HD)。HD不仅表现为舞蹈病,有时还表现为比舞蹈病本身更使人衰弱的认知和神经精神特征。患者以前被诊断出患有老年性舞蹈病,但不应将其作为最终诊断,而应寻找潜在病因,如HD、甲状腺功能亢进或自身免疫性原因。在HTT基因中具有少量扩展 CAG片段重复的个体通常具有迟发性表现,例如该患者。 |


一位36岁的妇女提供了一个评估她的进展性神经症状。在22岁时,她出现了全身性强直阵挛发作,需要多种抗癫痫药物来控制,包括拉莫三嗪、加巴喷丁和唑尼沙胺。有一次,她把开水泼到自己身上,寻求在烧伤科住院并植皮。32岁时,她出现了语言和吞咽问题。她说,她吃东西的时候舌头会不由自主地把食物往外推,而且她还经常呛噎,曾经需要海姆利克动作。她咬着嘴唇和舌头,每天24小时戴着咬合块。她的家人很难听懂她在电话里的讲话。在35岁的时候,她开始了手臂、躯干和腿部的剧烈颤动。她行走困难,多次摔倒。她在22岁左右患上了抑郁症、记忆力减退和易怒,但否认有强迫症行为。家族史阴性。 检查显示轻度帕金森病外观伴有面部肌张力障碍,尤其是面部下部区域。她有中度构音障碍。眼部检查显示垂直扫视的延迟启动,向下注视的范围比向上注视的范围更有限。她的四肢和躯干有轻微的间歇性舞蹈病,左臂也有一些肌张力障碍的姿势。她的步态复杂,部分由舞蹈病和肌张力障碍组成。她走路时左腿有些过度伸展和弯曲。深部腱反射消失。 外周血常规涂片中发现棘红细胞。她的肌酸激酶是790U/L。先前的脑部MRI报告显示无尾状体萎缩,但无法进行复查。她的血液样本Western blot显示没有舞蹈素蛋白。该患者确诊为舞蹈病-棘红细胞增多症。她的治疗由一个多学科团队管理,包括物理、职业和语言治疗师。 |
述评 舞蹈病,伴随摄食性肌张力障碍、癫痫发作以及合并神经病变或肌病(表现为深部腱反射消失或减少以及肌酸激酶升高),高度提示舞蹈病-棘红细胞增多症,这是一种常染色体隐性遗传的神经棘红细胞增多综合征。尽管在这个病人身上存在棘红细胞,但即便采用特殊技术,使用潮湿未固定的等渗稀释血液样本制备,在外周血涂片中也并不是总能见到棘红细胞。类似于亨廷顿病,眼跳延迟启动可见于舞蹈病-棘红细胞增多症;然而,与亨廷顿病相比,它通常出现在晚期。垂直性核上性眼肌麻痹是舞蹈病-棘红细胞增多症的一个不寻常的特征。舞蹈病类疾病的治疗并不局限于舞蹈症的治疗。虽然目前尚无具体的治疗方法,如果是轻微和不麻烦的舞蹈病,则不需要对症治疗,像这样的患者可以从物理疗法和康复专家以及物理、职业和语言治疗师的多学科治疗中受益。 与良性遗传性舞蹈病综合征相关的其他罕见基因在此仅作简要讨论。PDE10A突变可以是常染色体显性或隐性遗传。显性突变或二次突变通常在5-10岁左右表现为舞蹈病,在MRI上显示双侧纹状体高信号(因此,也称为儿童期发病型双侧纹状体坏死),但未见智力受损30。隐性遗传型更为严重。临床特征包括婴儿期发病的舞蹈症和运动语言发育迟缓,但有趣的是,MRI上并没有纹状体高信号31。 GNAO1基因功能获得性突变可表现为舞蹈病32,而功能丧失性突变可导致Ohtahara综合征,一这是种早期婴儿癫痫性脑病。与癫痫相关的舞蹈病中报告的相对较新的基因包括FOXG1(患者可能有明显的口面部舞蹈症/运动障碍)33、GRIN134-35和FRRS1L36。 |

| 疾病 | 遗传方式 | 基因(编码蛋白) | 选定的临床线索(舞蹈症除外) | 备注/注意事项 |
| 舞蹈病样脑瘫 | 未提及 | 未提及 | 肌张力障碍常与舞蹈症同时存在 (动作)的流畅成分有时很难区分肌张力障碍和舞蹈病手足徐动症 肌张力障碍性手部姿势通常不痛;健康人很难模仿 | 包含在这个表中,因为它可能是由遗传/神经代谢紊乱混合组成,除了缺氧性脑损伤,在作出这个诊断之前必须仔细排除。 |
| 良性遗传性舞蹈病综合征 | 可能是由于多种基因突变所致,包括NKX2-1、ADCY5、PDE10A、GNA01、SLC16A2(Allan-Herndon-Dudley综合征) | |||
| 经典型良性遗传性舞蹈病综合征或NKX2-1相关良性遗传性舞蹈病 | 常染色体显性遗传 | NKX2-1基因(以前称为TITF-1,甲状腺转录因子-1) | 也称为脑-肺-甲状腺综合征(或BLT综合征) 甲状腺功能减退和肺部疾病(如呼吸窘迫或间质性肺疾病)可以同时存在 典型的非进展性,但并非总是良性(与享廷顿病相比,认为相对更加良性) 患者可能发育迟缓或身材矮小 | 一些患者可能对左旋多巴治疗有矛盾的反应 |
| ADCY5相关的运动障碍 | 常染色体显性遗传 | ADCY5(腺苷酸环化酶5) | 表型多样性 混合性运动障碍,包括舞蹈症、肌张力障碍、肌阵挛运动可能是阵发性 | 以前被称为家族性运动障碍伴面肌抽搐,但根据肌电图检查,面部运动实际上并不是肌纤维抽搐 |
| Lesch-Nyhan综合征 | X连锁隐性遗传 | HPRT1(次黄嘌呤鸟嘌呤磷酸核糖转移酶) | 常伴有以下颅区域受累为主的肌张力障碍 自残行为 可能类似于舞蹈症-棘红细胞增多症,但Lesch-Nyhan综合征的发病年龄组通常更年轻 高尿酸血症 | 由于X染色体失活偏移女性患病的可能性较小 |
| Wilson病(肝豆状核变性) | 常染色体隐性遗传 | ATP7B(ATP7B) | 各种运动障碍,包括肌张力障碍、帕金森症、共济失调、震颤(包括典型的扑翼样震颤,一种小脑性震颤,但并不常见) Kayser-Fleischer环(注意上下角膜缘);如有疑问,参考裂隙灯检查 低血清铜蓝蛋白,低血清铜,高24小时尿铜 | |
| 常染色体隐性共济失调 | 大多数常染色体隐性共济失调起病于儿童期,可表现为多动性运动障碍,包括肌张力障碍、舞蹈症和肌阵挛 | |||
| Friedreich共济失调 | 常染色体隐性遗传 | 由GAA重复扩增和/或突变引起的FXN(frataxin) | 肌张力障碍、共济失调、高足弓、反射减退、糖尿病、心肌病、脊柱侧凸 眼球运动检查:巨视振荡,超视距扫视 在MRI 上相对保留小脑的大小直到晚期 变型(该二者重叠):Friedreich共济失调伴反射保留或反射亢进,常迟发;迟发性Friedreich共济失调 | |
| 共济失调伴1型动眼神经失用症(A0A1) | 常染色体隐性遗传 | APTX(aprataxin) | 动眼神经失用症(眼跳延迟开始) 神经病变 低血清白蛋白,高胆固醇 | 与共济失调相关的动眼神经失用症的鉴别诊断包括共济失调毛细血管扩张症、A0A1和A0A2 |
| 共济失调伴2型动眼神经失用症(A0A2) | 常染色体隐性遗传 | SETX(番泻叶毒素) | 年龄组通常比A0A1大 动眼神经失用症 神经病变 几乎所有病例都有α-甲胎蛋白(AFP)升高 | 在共济失调-毛细血管扩张症(几乎所有病例)中,α-甲胎蛋白也会升高 |
| 代谢紊乱(例如有机酸血症) | 常染色体隐性遗传 | 多种 | 也可出现肌张力障碍 双侧豆状核T2加权MRI上的高信号 | 鉴别诊断包括甲基丙二酸血症和戊二酸尿症I型等 检查血浆氨基酸和尿有机酸 |
| 线粒体疾病(如Leigh 综合征或MELAS) | 线粒体或常染色体隐性遗传(如果归因于核突变) | 线粒体/核基因突变 | 也可能出现肌张力障碍 Leigh综合征的MRI表现:T2加权序列双侧基底节和/或脑干高信号 MELAS可有基底节钙化(CT有助于显示) | 首先检查血清和脑脊液中的高乳酸水平 |
一名6岁男孩来到诊所对他的舞蹈病进行随访。他足月出生,没有任何产前和围产期并发症。然而,在生命的前两年,他被诊断为甲状腺功能减退和“哮喘”。大约4.5岁时,他在运动障碍门诊接受了评估,因为在过去的1.5年中,伴随着全身性舞蹈症的异常运动逐渐变得更加明显。母方的多个家庭成员有甲状腺问题和哮喘;然而,这些家庭成员没有异常运动的报告。诊断良性遗传性舞蹈症的是可疑的,并证实了存在NKX2-1(以前称为TITF-1)基因突变。他有轻度智力障碍,曾参加过一个特殊的学校项目。他的舞蹈症在过去的两年里一直很稳定。他在6岁时接受左旋多巴试验治疗,最高剂量为5mg/kg/d,以控制舞蹈症的症状。但是,由于没有看到任何改善,因此后来停止了。 检查显示轻中度全身性舞蹈症,包括所有四肢、躯干、颈部和面部下部,以及行走时表现为轻度足内翻的双侧轻度足肌张力障碍。与舞蹈症相比,肌张力障碍不太明显。治疗方案包括丁苯那嗪,与他的父母进行了讨论。 |
述评 这名患者的慢性时间变化谱提示舞蹈病的遗传原因,其中最常见于儿童的是良性遗传性舞蹈病。良性遗传性舞蹈病综合征可由多种基因突变引起。“经典型”良性遗传性舞蹈病综合征是由NKX2-1(TITF-1)突变引起的,这种突变也会引起肺和甲状腺的问题(因此称为脑-肺-甲状腺综合征),正如该患者。全部三个器官的受累只出现在30-40%的患者,而在该患者身上可以见到。良性遗传性舞蹈病并不总是“良性的”;例如,该患者有智力障碍。某些,但并非所有,患有该疾病的患者可以对左旋多巴有反应。 |
| 疾病 | 遗传方式 | 基因(蛋白质编码) |
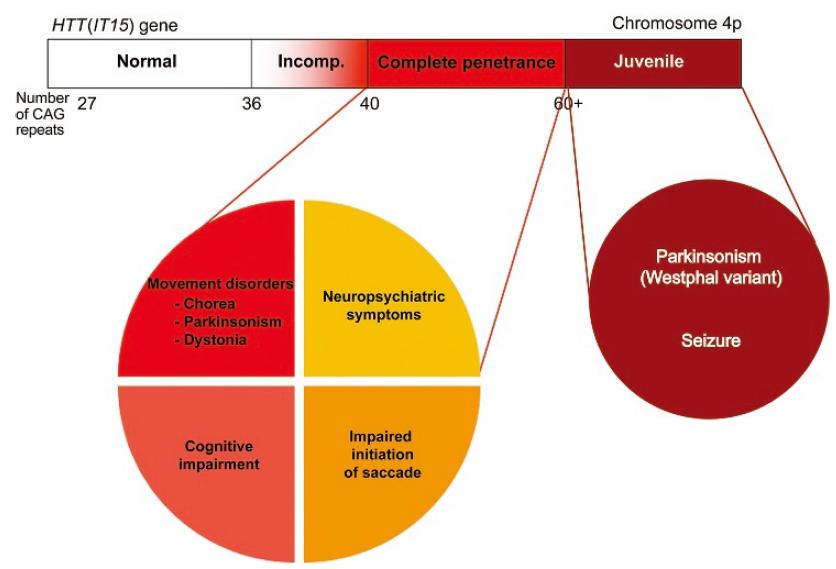
| 亨廷顿舞蹈病 | 常染色体显性遗传 | HTT(也称为IT15)基因(亨廷顿蛋白)中CAG重复扩增36-39次: 外显率降低 ≥40:完全外显率 >60:青少年亨廷顿病(威斯特法尔变异型) |
| c9orf72 疾病 | 常染色体显性遗传 | GGGGCC在C9orf72基因中的重复扩增(C9orf72) |
| SCA17(HDL-4) | 常染色体显性遗传 | TBP(TATA-盒结合蛋白) |
| 亨廷顿病-2(HDL-2) | 常染色体显性遗传 | CTG/CAG 在 JPH3 亲子蛋白中的重复扩增 |
| 神经棘红细胞增多症 | ||
| 棘红细胞增多症-舞蹈病 | 常染色体隐性遗传 | VPS13A (舞蹈素) |
| McLeod 综合征 | X连锁遗传 | XK (Kx 抗原) |
| 齿状-苍白萎缩 | 常染色体显性遗传 | ATN1(阿托芬 1) |
| 神经变性伴脑铁蓄积 | ||
| 神经铁蛋白病 | 常染色体显性遗传 | FTL (铁蛋白轻链,ferritin light chain) |
| 血浆铜蓝蛋白缺乏症 | 常染色体隐性遗传 | CP (血浆铜蓝蛋白,ceruloplasmin) |
登录后 PLUS 会员 可查看完整内容

微信扫一扫
成为会员后
查看完整内容
文献评审日期:2021-05-31
1. Termsarasab P. Hyperkinetic movement disorders: videodiagnosis and treatment. Presented at:69th Annual Meeting of the American Academy of Neurology; April 22–28, 2017; Boston, MA.
3. Varley JA, Webb AJS, Balint B, et al. The movement disorder associated with NMDAR antibody-encephalitis is complex and characteristic: an expert video-rating study. J Neurol Neurosurg Psychiatry 2018;pii:jnnp-2018–318584. doi:10.1136/jnnp-2018-318584.
感谢以下医疗从业者参与贡献
排序不分先后