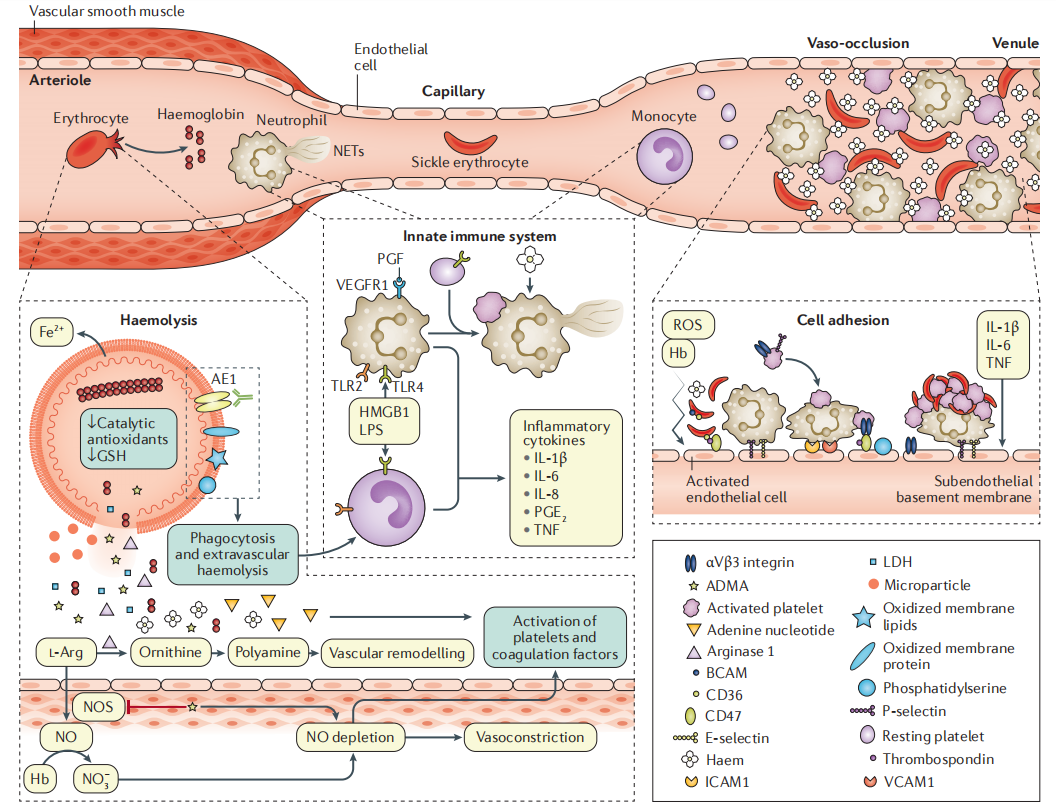
(2)血浆游离血红蛋白和血红素(血浆或微粒中的[27-28])细胞外血红蛋白和血浆血红素会促进严重氧化应激的发生,尤其对血管和血细胞[28]。细胞外血红蛋白持续自氧化生成超氧化物,超氧化物又分解成过氧化氢(hydrogen peroxide,H2O2),而过氧化氢是其他强效氧化活性物质的来源,包括促进血管收缩的高价铁离子[28]。细胞外血红蛋白清除一氧化氮[nitric oxide,NO;由内皮细胞 NO 合成酶(NO synthase,NOS)生成,可促进血管舒张]的速度约为细胞质血红蛋白的 1000 倍,从而降低了 NO 的生物利用度[38]。NO 生物利用度降低导致血管功能障碍,表现为血管对 NO 供体舒张反应受损、内皮细胞激活(产生内皮粘附分子的细胞表面表达,通过粘附分子溶于血浆的胞外域的表达检测出来)和血小板止血功能激活,而血小板止血功能激活表现为细胞表面 P-选择素表达(介导活化的血小板和白细胞相互作用)和 αIIbβ3 整联蛋白活化[33]。溶血严重程度的标志物(如低血红蛋白或高血清乳酸脱氢酶)可预测发生血管疾病并发症的临床风险。
(3)精氨酸代谢紊乱
血管内溶血释放两个干扰 NOS 活性的因子。精氨酸酶 1 与 NOS 竞争 L-精氨酸,而 L-精氨酸是 NOS 生成 NO 所需的底物[39]。精氨酸酶 1 将 L-精氨酸转化为鸟氨酸,鸟氨酸可促进多胺的合成,进而促进细胞增殖[40],增殖的细胞可能为血管细胞,这可能会促进血管重建。不对称二甲基精氨酸(asymmetric dimethylarginine,ADMA)是一种内源性 NOS 抑制剂,也是精氨酸甲基化蛋白的水解产物;红细胞含大量 ADMA,在溶血过程中也会释放 ADMA[41]。ADMA 和精氨酸酶 1 耗竭 L-精氨酸均有助于 NOS 解偶联,随后 NOS 生成活性氧(reactive oxygen species,ROS)而非 NO[42-43]。
(4)血脂
SCA 患者通常有一种与血管病变相关的血脂异常:甘油三酯水平升高,且与溶血严重程度相关[44]。尽管 SCA 患者总胆固醇水平通常较低,但载脂蛋白 A-I(促进肝脏胆固醇分解代谢,增强 NOS 活性)水平特别低,尤其是在血管闭塞性疼痛危象期间,并与肺动脉高压和内皮功能障碍的标志物相关[45]。载脂蛋白 L 1 基因突变与 SCA 中的肾脏疾病有关[46]。
5、固有免疫系统激活血浆血红素和血红蛋白作为损伤相关分子模式(danger-associated molecular patterns,DAMPs)激活固有免疫系统,加强循环血细胞彼此之间及与内皮的粘附,从而触发血管闭塞[33](图 3)。血红素激活中性粒细胞,使其释放 DNA,形成中性粒细胞胞外陷阱(neutrophil extracellular traps,NETs),NETs 增加血小板活化,加速血栓形成,促进肺血管闭塞[47]和有核红细胞(红细胞的有核前体)释放胎盘生长因子(placenta growth factor,PGF)。PGF 是内皮细胞和巨噬细胞上血管内皮生长因子受体 1 的配体,促使细胞释放内皮素 1(一种血管收缩剂),导致肺动脉高压[48]。Toll 样受体 4(Toll-like receptor 4,TLR4)在 SCD 免疫细胞中高表达,组织损伤和血小板激活释放一种高亲和力的 TLR4 配体——高迁移率族蛋白 B 1(high mobility group protein B 1,HMGB1)。TLR4 还可结合来源于革兰氏阴性菌的脂多糖(lipopolysaccharide,LPS),这可解释为什么感染会促使 SCA 患者发生血管闭塞危象。TLR4 配体激活单核细胞和巨噬细胞,使其释放炎性细胞因子,促进炎症状态,激活中性粒细胞、血小板和内皮细胞的黏附。最后,反复溶血和输注红细胞导致的细胞内铁增多与多种炎症小体通路成分在外周血单核细胞中表达显著增加相关[49]。